
{kind=link}
While it is possible today to measure in such rupture experiment the forces as the molecule is stretched, it is often difficult to infer directly from experiment what happens on the atomic scale. Due to dramatic developments in computer simulation techniques and in particular due to the so-called Car-Parrinello molecular dynamics method, it is possible to perform first principles or ab initio calculations of such processes in the “virtual laboratory”, i.e. a computer fed with an appropriate computer program. These simulations do not rely on experimental input or parameters, but are solely based on the basic laws of physics in conjunction with well-defined approximations and numerical tricks.
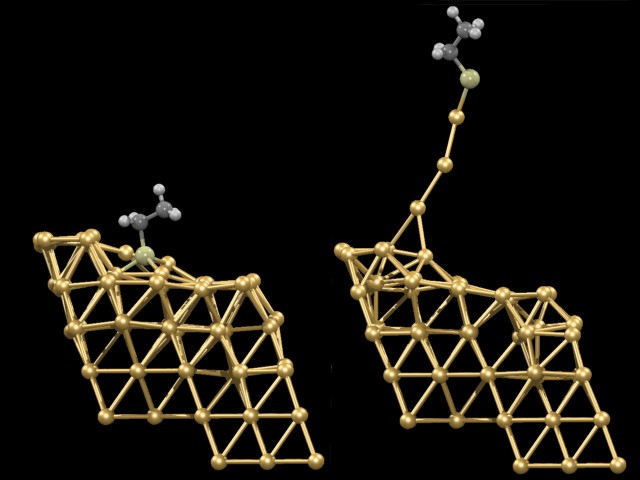
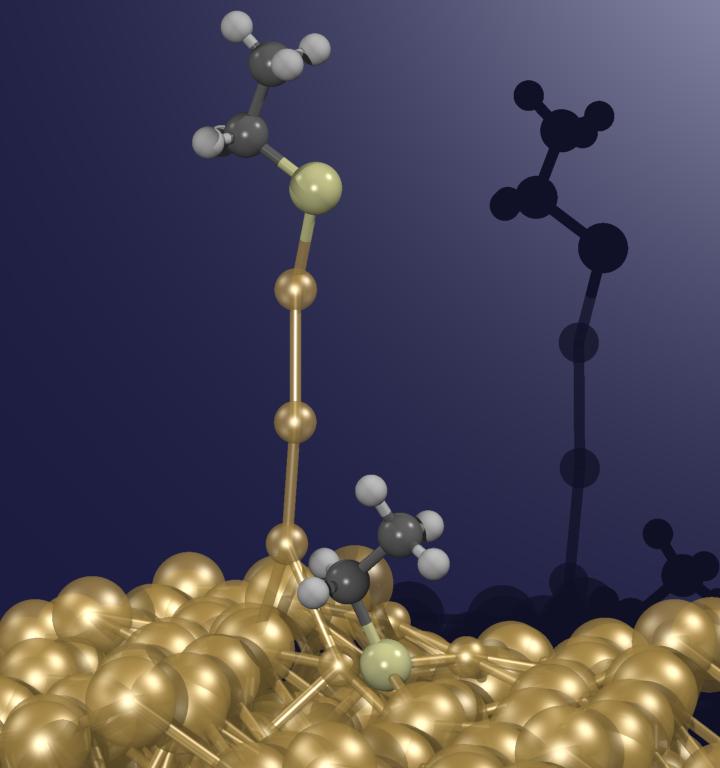
Using such techniques, it was investigated what happens if one particular molecule (ethylthiolate) is first anchored on a gold surface (an Au(221) surface was selected as an Au(111) vicinal surface) and subsequently pulled off the surface by exerting an external force on the terminal C-atom perpendicular to the Au(111) surface. From the animation it can be seen that the gold surface does not remain unperturbed: very large atomic rearrangements take place as the molecule is absorbed and in particular during the pulling process. What is most astonishing is that gold atoms are extracted out of the surface. These gold atoms finally form an “atomic necklace”, i.e. a monoatomic gold nanowire. After having extracted about three to four atoms the wire suddenly breaks. A linear chain of three gold atoms remains attached to the molecule but “isomerizes” instantaneously to a more stable triangular gold cluster. Thus it is the gold-gold bond that connected the nanowire to the surface that breaks preferentially, and not the contact junction between the molecule and the gold surface (which is a sulfur-gold bond). The reason is that the junction gets actually strengthened in a synergistic manner because as the pulling process occurs sulfur trades three weak bonds to gold for one very strong one. In summary, this computer experiment yielded an ultimately detailed atomistic view on what happens as a molecule gets detached from a metal surface.

The behavior of copper, another coinage metal, is very different from that of gold when it comes to thiolate/metal junctions and interfaces. In particular, it is found that thiolate/copper junctions have a much stronger preferrence to break the C-S bond within the molecule in comparison to the analogues gold systems. This matches well with experimental observation of the thermal desorption of hydrocarbons from self-assembled thiolate monolayers on bulk copper surfaces where sulfur remains on the surface. On the other hand, for gold, relativistic contributions to the bonding lead to a weaker and more covalent metal-thiolate interaction where S-C bond weakening is mitigated. This thus is a “relativistic effect”: hypothetical non-relativistic gold behaves similar to copper with an even more pronounced propensity for S-C bond rupture.
Significant differences are also observed when is comes to thiolate/metal interfaces. In stark constrast to CH3S/Au(111), the flat adsorption energy landscape of CH3S/Cu(111 results from metal-to-molecule charge redistribution and non-directional, partially ionic bonding, and to polarization effects within the molecules forming the self-assembled monolayer. Alltogether this allows for an astonishingly high lateral mobility of the molecules on the surface (and thus leads to a significant non–uniform diffuse background to Bragg scattering which must be taken properly into account when analyzing experimental diffraction data). Furthermore, these findings of charge redistribution and molecular polarization suggest that the properties of such molecule/metal interfaces may be manipulated by tuning the chemical potential of the organic molecules used, e.g. by suitable chemical substitution of the thiolates, relative to the work function of the underlying metal surface.
Concerning the calculation itself, it should be noted that such large-scale simulations require a huge amount of supercomputer resources. Most of those were provided by the Scientific Supercomputer Center Karlsruhe (SSCK) on its IBM RS/6000 SP. The SSCK/HLRS Steering Committee honored Daniel Krüger with the “Golden Spike Award 2001”, which is awarded to the three most outstanding projects in each year.
Further information and details:
- Quantum simulations (constrained canonical Car-Parrinello molecular dynamics)
- Daniel Krüger (Westfälische Wilhelms-Universität Münster)
- Roger Rousseau (Ruhr-Universität Bochum)
- Martin Konôpka (Slovak University of Technology Bratislava)
in collaboration with - Harald Fuchs (Westfälische Wilhelms-Universität Münster)
- Michele Parrinello (Swiss Center for Scientific Computing/ETH Zurich)
- Ivan Štich (Slovak University of Technology Bratislava)
- Dominik Marx (Ruhr-Universität Bochum)
- Scientific visualization
- Daniel Krüger (Westfälische Wilhelms-Universität Münster)
- Additional information
- RUB Pressemitteilung
- Dynamics of self-assembled thiolate monolayers on copper:
M. Konôpka, R. Rousseau, I. Štich, and D. Marx,
Electronic Origin of Disorder an Diffusion at a Molecule-Metal Interface:
Self-Assembled Monolayers of CH3 on Cu(111)
Phys. Rev. Lett. 95, 096102 (2005) - Mechanochemical behavior of moelcule/metal juntions: copper versus gold:
M. Konôpka, R. Rousseau, I. Štich, and D. Marx,
Detaching Thiolates from Copper and Gold Clusters:
Which Bond to Break?
J. Am. Chem. Soc. 126, 1203 (2004) - Gold-thiolate cluster AFM calculations:
D. Krüger, H. Fuchs, R. Rousseau, and D. Marx,
Towards “Mechanochemistry”: Mechanically Induced Isomerization of Thiolate-Gold Clusters,
Angew. Chem. Int. Ed. 42, 2251–2253 (2003) and cover picture.
Auf dem Weg zur „Mechano-Chemie“: mechanisch induzierte Isomerisierungen von Thiolat-Goldclustern,
Angew. Chem. 115, 2353–2355 (2003) und Titelbild. - Gold-thiolate surface AFM calculations:
D. Krüger, H. Fuchs, R. Rousseau, D. Marx, and M. Parrinello,
Pulling monatomic gold wires with single molecules: An ab initio simulation,
Phys. Rev. Lett. 89, 186402 (2002) - Golden Spike Award 2001
- Gold-thiolate cluster calculations:
D. Krüger, H. Fuchs, R. Rousseau, D. Marx, and M. Parrinello,
Interaction of short-chain alkane thiols and thiolates with small gold clusters: Adsorption structures and energetics,
J. Chem. Phys. 115, 4776–4786 (2001) - Gold-ethanol cluster calculations:
G. M. Koretsky, M. B. Knickelbein, R. Rousseau, and D. Marx,
A Combined Infrared Photodissociation and Theoretical Study of the Interaction of Ethanol with Small Gold Clusters,
J. Phys. Chem. A 105, 11 197–11 203 (2001) - Gold-methanol cluster calculations:
R. Rousseau, G. Dietrich, S. Krückeberg, K. Lützenkirchen, D. Marx, L. Schweikhard, and C. Walther,
Probing cluster structures with sensor molecules: methanol adsorbed onto gold clusters,
Chem. Phys. Lett. 295, 41–46 (1998)
R. Rousseau and D. Marx,
The interaction of gold clusters with methanol molecules: Ab initio molecular dynamics of Aun+CH3OH and AunCH3OH,
J. Chem. Phys. 112, 761–769 (2000) - Expert introduction into the simulation techniques
- This web page
http://www.theochem.ruhr-uni-bochum.de/go/afm.html